MLZ ist eine Kooperation aus:
MLZ ist Mitglied in:
 > ERF-AISBL
> ERF-AISBL
MLZ in den sozialen Medien:


MLZ
Lichtenbergstr.1
85748 Garching
On the complex H-bonding network in paravauxite, Fe2+Al2(PO4)2(OH)2·8H2O
G. D. Gatta1, P. Vignola1,2, and M. Meven3,4
1Dipartimento di Scienze della Terra, Università degli Studi di Milano, Milano, Italy
2CNR-Istituto per la Dinamica dei Processi Ambientali, Milano, Italy
3Institut für Kristallographie, RWTH Aachen, Aachen, Germany
4Jülich Centre for Neutron Science (JCNS) at MLZ, Forschungszentrum Jülich GmbH, Garching, Germany
Phosphate minerals represent the major host for transition metals and H2O in pegmatitic rocks, playing an essential geochemical role in the evolution processes of pegmatites. A good knowledge of their crystal chemistry is therefore necessary to better understand the genesis of pegmatites. Paravauxite is a mineral found in hydrothermal tin veins and granite pegmatites [1,2]. Its ideal chemical formula is Fe2+Al2(PO4)2(OH)2·8H2O. Its crystal structure was solved and refined by Baur [3] in 1969 on the basis of single-crystal X-ray diffraction data. This structure model appears to be consistent. However, due to the technical limitations of X-ray diffraction, the refinement only provided the isotropic displacement parameters, and the positions of nine independent proton sites were assigned but not refined. This led to a poor description of (the expected) complex H-bonding scheme in the paravauxite structure. In light of this, the crystal structure of a natural paravauxite was reinvestigated using electron microprobe analysis in wavelength dispersive mode (EPMA-WDS) and single-crystal neutron diffraction in an attempt to resolve these open questions.
Looking into a gem stone
A gemmy, pale green, single crystal of paravauxite (up to 9 mm in length and 5 mm in diameter) from the Siglo Veinte Mine, Bolivia, was used in this study. The determination of the chemical composition was performed by EPMA-WDS analysis on a polished crystal using a Jeol JXA-8200 microprobe with the following result:
Fe(Fe2+0.916Mn2+0.016Mg0.064Ca0.002)Σ0.998Al(1)Al(2)Al2.005P(P1.998Si0.002)Σ2O8(OH)2·8H2O.
A single-crystal neutron diffraction experiment was performed using the hot source (fast neutrons) single-crystal diffractometer HEiDi of the neutron source FRM II. The diffraction data were collected at 293 K with a wavelength of the incident beam of 1.1680(2) Å. The unit-cell parameters were refined on the basis of the 42 Bragg reflections (space group: P -1, a = 5.240(6) Å, b = 10.567(7) Å, c = 6.698(9) Å, α = 106.82(8)°, β = 110.77(9)°, γ = 72.23(9)°, V = 336.4(6) Å3). A total number of 4190 reflections were collected up to 2θmax = 126.3° and sin(Θ)/λ = 0.76/Å, respectively. The discrepancy factor for the symmetry related reflections (based on Friedel pairs) was Rint = 0.0442. The anisotropic structure refinement was then performed using the SHELX-97 software [4], starting from the atomic coordinates of Baur [3] without H sites. The structure refinement was conducted with: a) the neutron scattering length of iron at the octahedral Fe site and the scattering length of aluminum at the octahedral Al(1) and Al(2) sites, also refining their site occupancy factors (s.o.f.); b) the scattering length of phosphorous at the tetrahedral P site, with full occupancy; c) the scattering length of oxygen at the OP(1), OP(2), OP(3), OP(4), OH(5), OW(6), OW(7), OW(8) and OW(9), with full site occupancies. Then, a structure model was implemented with nine H sites, (i.e., H(1), H(2), H(3), H(4), H(5), H(6), H(7), H(8) and H(9)) all at about 1 Å from the respective O sites. Given such a model, convergence was rapidly achieved. However, H(4) and H(9) showed unrealistically large displacement parameters, if compared to those of the other H sites. Further refinement cycles were then conducted splitting the H(4) and H(9) sites into two mutually exclusive sub-sites (i.e., H(4A) and H(4B), H(9A) and H(9B)) only 0.4-0.6 Å apart. Their s.o.f. were not restrained. With this configuration, the refined displacement parameters had realistic values, convergence was achieved and the variance-covariance matrix showed no significant correlation among the refined parameters. No peak larger than ±1.3 fm/Å3 was present in the final difference-Fourier map of the nuclear density. The final statistical index R1 was 0.0495 for 194 refined parameters and 1678 unique reflections with Fo > 4σ(Fo).
Locating the hydrogen in paravauxite
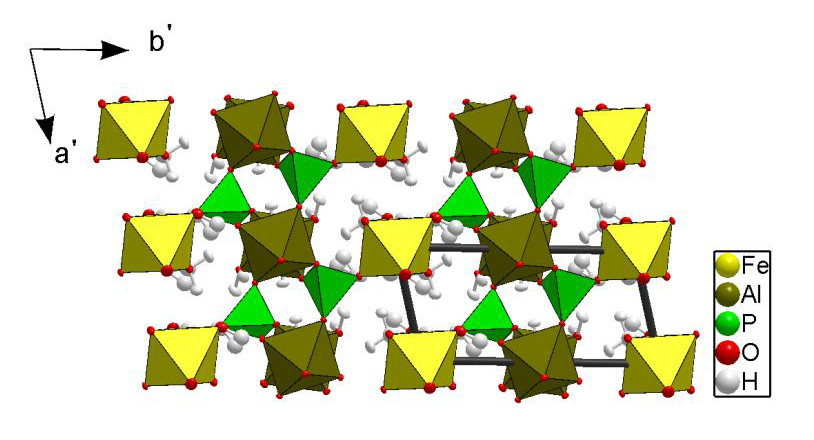
This is the first study in which the crystal structure of paravauxite has been investigated on the basis of single-crystal neutron diffraction. Previous structure data available in the literature [3] are based on single-crystal X-ray diffraction. The structural refinement of this study confirms the former general structure model [3]. The structure of paravauxite is composed of chains of corner-sharing Al-octahedra, running along [001], linked by P-tetrahedra to form layers parallel to the ac-plane. These layers are connected by Fe-octahedra (Fig. 1). Two independent Al-octahedra (i.e., AlO4(OH)2 and AlO2(OH)2(OH2)2), one independent Fe-octahedron (i.e., FeO4(OH2)2) along with one independent PO4-tetrahedron form the polyhedral “framework”, and at least one independent “zeolitic” H2O lies in the cavities.
Using the neutron scattering length of iron at the Fe site, the refined occupancy factor is s.o.f. = 0.921(7). This virtual partial site occupancy reflects the multi-elemental population at the Fe site, as shown by the EPMA-WDS [i.e., with minor fractions of Mg (0.064 a.p.f.u.) and Mn (0.016 a.p.f.u.)]. The Al(1) and Al(2) sites were found to be fully occupied by aluminum (with refined s.o.f. = 1.02(2) and 1.05(2), respectively). The s.o.f. of the subsites H(4A) and H(4B), and H(9A) and H(9B) were refined without any restraint, and the sum [s.o.f.(H4A) + s.o.f.(H4B)] = 0.94(3) and [s.o.f.(H4A) + s.o.f.(H4B)] = 1.02(2) suggest full site occupancies within 2σ. The structure model with the sub-sites H(4A) and H(4B), and H(9A) and H(9B) is the best fit to the observed intensity data (at 293 K), with realistic displacement parameters.
The complex H-bonding scheme in the paravauxite structure is now well defined, with twelve independent H-bonds. Some of the H-bonds appear to be stronger than others. The weaker are characterized by low O-H⋅⋅⋅O angular values (i.e., 123 – 146°). Some H-bonds connect the Al-octahedra with the Fe-octahedra. The zeolitic H2O molecule (i.e., H(8)-OW(9)-H(9AB)) is connected via H-bonding to OP(1) (i.e., the bridging oxygen between the Al(1)-octahedron and the P-tetrahedron), OP(3) (i.e., the bridging oxygen between the Fe-octahedron and the P-tetrahedron) and OW(6) (i.e., belonging to the Al(2)-octahedron). Further structural details are reported in [5].
References:
[1] S. G. Gordon, Proc. Acad. Nat. Sci. Philadelphia 75, 261 (1922).
[2] S. G. Gordon, Proc. Acad. Nat. Sci. Philadelphia 96, 279 (1944).
[3] W. H. Baur, Neues Jahrbuch für Mineralogie Monatshefte 1969
430 (1969).
[4] G.M. Sheldrick, Acta Crystallogr. Sect. A 64, 112 (2008).
[5] G. D. Gatta et al., Mineralogical Magazine 78, 841 (2014).
MLZ ist eine Kooperation aus:
MLZ ist Mitglied in:
> ERF-AISBL
MLZ in den sozialen Medien: